Computational Biology Unit
We are interested in developing algorithms, models and tools for the analysis of genomic data that enable researchers to understand what biological processes or variants are involved in differentphenotypes or diseases. For doing that we apply techniques, concepts and analysis of Scientific Computing, which is the subfield of computer science concerned with constructing mathematical models and quantitative analysis techniques and using computers to analyze and solve scientific problems.
CBU's main lines of reasearch are:
- Discovery, characterization and analysis of genomic variants: Understanding the relationship between genomic variants and phenotype is one of the main goals in biology and medicine. New findings show that genomic variants are more common than expected making more difficult the analysis and interpretation of results. We are interested in characterizing and developing new methods and algorithms to help researchers to study these genomic variants. These methods range from statistical tests to prioritization or biological network-based algorithms.
- Biological networks and Systems Biology modeling: Genes, proteins and regulatory elements operate within an intricate network of interactions. A new paradigm has emerged to study these biological systems, this new holistic paradigm aims to understand how the interactions of the components of biological systems give rise to the function and how they participate in penotypes and diseases. We are interested in developing new algorithms and tools to model and analize these biological networks.
- Software development for data analysis: Recent advances in high-throughput technologies such as NGS make harder than ever the analysis of genomic data because of the size and heterogeneity of data. Today the bottleneck is no data generation but data analysis, data is generated in a few hours or days, but analysis can take weeks or months. We need new computing solutions to allow researchers to work with this huge volume of data efficiently and to work in a distributed enviroment like in a cloud. We are developing DNA and RNA aligners and solutions to analize with genomic variants.
- Integrative information system and WEB services: During last years the number of biological databases has grown exponentially. Today biological information is spread out over more than 1000 databases making dificult the retrieval, integration and access to the data as these databases use different standards. We are developing a centralized database with the most useful biological information from different sources and making all these information accessible through WEB services, by doing so biological information will be available easily to researchers to accelerate data analysis.
To achieve this goals we use advanced computing solutions:
- WEB applications and cloud-based solutions for data analysis: The size of the data currently produced by new high-throughput technologies such as NGS force us to think new ways to store and analyze genomic data. The incredible size of new experiments, up to some TeraBytes, make it impossible to store in curent workstations or even move the data over the network. We are exploring and developing new strategies and WEB applications to efficently store, analyze and explore data in a cloud.
- HPC software development for data analysis: Recent advances in high-throughput technologies such as NGS make harder than ever the analysis of genomic data because of the size and heterogeneity of data. We are developing a next generation software in Bioinformatics using HPC computing that exploits the current hardware and computing technologies such as multi-core CPUs or GPGPUs. This software aims to implement most useful analysis on a minute scale without sacrificing any feature.
- Machine learning software development: Recent years we have seen how many of the high-throughput techniques like expression microarrays are being used in clinics to develop diagnostic tools, during last years FDA has approved many of these predictors. Machine learning algorithms can be organized into supervised learning (SVM, KNN, Neural networks, ...) and unsupervised learning (clustering and data mining). We are using this Machine learning algorithms to build predictors based on expression data, and to knowledge discovery or find patterns in data.
Technologies
To develop our solutions we use the most modern computer technologies available:
- WEB applications and WEB services: We use modern HTML5 technologies for building rich client applications and RESTful WEB services to make data and analysis available in a efficient way from our servers.
- HPC technologies: We combine OpenMP and SSE/AVX instructions of CPU to implement efficiently algorithms or analysis. We also have used some Nvidia CUDA software implementation when appropiate.
- Distributed and Cloud computing: We use Apache Hadoop framework to analize with MapReduce and store large volumes of data with NoSQL HBase. We are also using Amazon AWS to export some of our services.
- Machine learning and Computational modeling: We use machine learning algorithms such as predictors or clustering to solve some of the problems when looking for patterns in data. We also use graph theory to model biological networks and develop methods for the analysis.
WEB applications
Babelomics is an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. It is free and can be found at http://babelomics.bioinfo.cipf.es.
Genome Maps has been built using HTML5 technology available in modern WEB browsers. Genome Maps is designed to provide a real-time navigation and allow researchers to better analyze their data. Genome Maps data is stored in a MySQL cluster that export data through a complete RESTful WEB service API: http://www.genomemaps.org.
CellBase is an ambitious project in which we have integrated many different sources of biological information together and we have made all this information available through a RESTful WEB service APIS. More information can be found at http://docs.bioinfo.cipf.es/projects/cellbase.
Others WEB applications are VARIANT for VCF genomic variants analysis (http://variant.bioinfo.cipf.es) and RENATO for the interpreatation and visualization of transcriptional and post-transcriptional regulatory information (http://renato.bioinfo.cipf.es).
HPC solutions
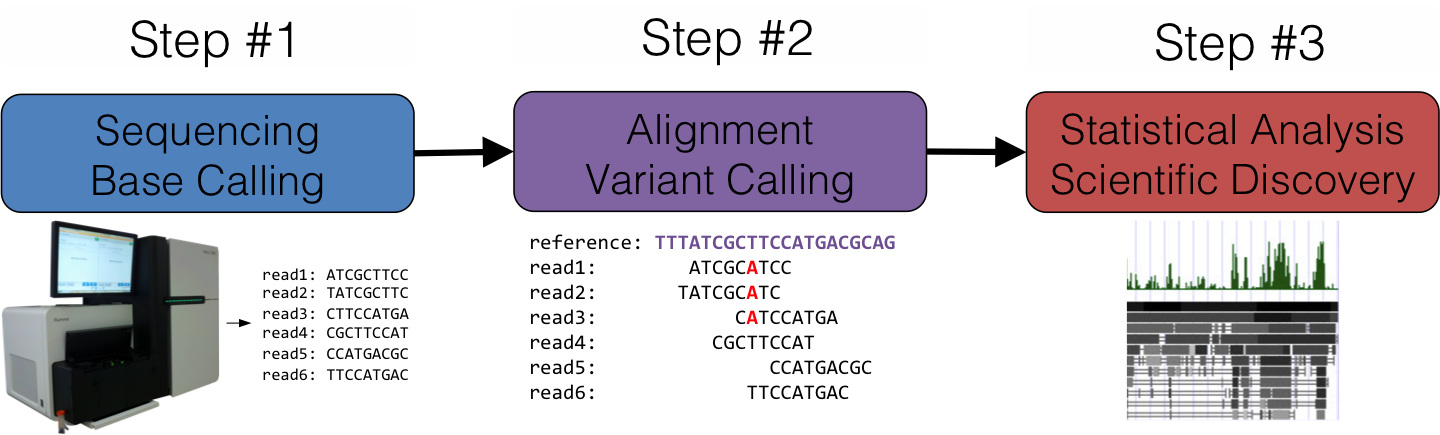
DNA short read aligner has been developed using a combined strategy of Burrows-Wheeler Transform and Smith-Waterman algorithms. Implementation has been carried out using HPC technologies to speed-up performance. Results show an amazing sensibility detecting indels and any number of mismatches.
RNA-seq aligner has been developed following a similar approach, using a combined strategy of Burrows-Wheeler Transform and Smith-Waterman algorithms. Implementation has been carried out using HPC technologies to speed-up performance. First results show more than 90% of reads mapped and high sensibility detecting splice junctions.
HPG-Variant a complete suite to work with VCF files. It is implemented HPC techniques to speed-up performance. Many gwas analysis and utilities are being implemented.
HPG-Fastq tool implements usign OpenMP and Nvidia CUDA technolgies any useful utilities to work with huge Fastq files efficiently.
Smith-Waterman HPC C library and CLI application have been implemented using HPC technologies such as OpenMP and SIMD SSE instructions providing a speed up of ~32x over other implementations in a octa-core processor.