High Performance Genomics for NGS data
- HPG Aligner: Ultrafast and highly sensitive Next-Generation Sequencing (NGS) read mapping
- HPG Pore: Toolkit for exploring and analysing nanopore sequencing data human
- ngsCAT: a tool to assess the efficiency of targeted enrichment sequencing
Variant annotation
Variant Visualization
- GenomeMaps: Our higly efficient nex generation genome viewer (used by the ICGC)
Variant prioritization
- BiERapp: Variant/gene prioritization tool (from VCF to candidates)
Diagnostic
- TEAM: Targeted enrichment sequencing (panels) diagnostic tool
Transcriptional networks
- RENATO Inferring the regulatory network behind a gene expression experiment
Network analysis
- Snow Network enrichment of a set of genes
- Network Miner: Network enrichment analysis of a ranked list of genes
Pathway analysis
Packages
- Babelomics A program suite for advanced functional genomic data analysis
Tools no longer supported
Tools discontinued
- GEPAS Microarray data analysis
NGS
VARIANT: Command Line, Web service, and Web interface for fast and accurate functional characterization of variants found by Next Generation Sequencing
The massive use of Next Generation Sequencing (NGS) technologies is uncovering an unexpected amount of variability. The functional characterization of such variability, particularly in the most common form of variation found, the Single Nucleotide Variants (SNVs), has become a priority that needs to be addressed in a systematic way. VARIANT (VARIant ANalyis Tool) reports information on the variants found that include consequence type and annotations taken from different databases and repositories (SNPs and variants from dbSNP and 1000 genomes, and disease-related variants from the GWAS catalog, OMIM, COSMIC mutations, etc.) VARIANT also produces a rich variety of annotations that include information on the regulatory (transcription factor or miRNA binding sites, etc.) or structural roles, or on the selective pressures on the sites affected by the variation. This information allows extending the conventional reports beyond the coding regions and expands the knowledge on the contribution of non-coding or synonymous variants to the phenotype studied. Contrarily to other tools, VARIANT uses a remote database and operates through efficient RESTful Web Services that optimize search and transaction operations. In this way, local problems of installation, update or disk size limitations are overcome without the need of sacrifice speed (thousands of variants are processed per minute).
Implementation
VARIANT is available at: http://variant.bioinfo.cipf.es.
Publication
Medina I, De Maria A, Bleda M, Salavert F, Alonso R, Gonzalez CY, Dopazo J. 2012 VARIANT: Command Line, Web service and Web interface for fast and accurate functional characterization of variants found by Next-Generation Sequencing. Nucleic Acids Res. 40(Web Server issue):W54-8.
VARIANT is quoted in GenomeWeb and The Bulletin Electronique (discontinued).
CellBase: a comprehensive collection of RESTful Web Services for retrieving relevant biological information from heterogeneous sources.
During the last years, the advances in high-throughput technologies have produced an unprecedented growth in the number and size of repositories and databases storing relevant biological data. Today, there is more biological information than ever but, unfortunately, the current status of many of these repositories is far from being optimal. Some of the most common problems are that the information is spread out in many small databases, frequently there are different standards among repositories, some databases are not longer supported or they contain too specific and unconnected information. In addition, data size is increasingly becoming an obstacle when accessing or storing biological data. All these issues make very difficult to extract and integrate information from different sources, to analyze experiments or to access and query this information in a programmatic way. CellBase provides a solution to the growing necessity of integration by easing the access to biological data. CellBase implements a set of RESTful Web services which query a centralized database containing the most relevant biological data sources. The database is hosted in our servers and is regularly updated.
Publication
Genome Maps a new generation genome browser
Genome browsers have gained importance as more genomes and related genomic information be-come available. However, the increase of information brought about by new generation sequencing technologies is, at the same time, causing a subtle but continuous decrease in the efficiency of conventional genome browsers. Here we present Genome Maps, a genome browser that implements an innovative model of data transfer and management. The program uses highly efficient technologies from the new HTML5 standard, such as SVG, that optimize workloads at both, server, and client sides and ensure future scalability. Thus, data management and representation are entirely carried out by the browser, without the need of any Java Applet, Flash or other plug-in technology installation. Relevant biological data on genes, transcripts, exons, regulatory features, single nucleotide polymorphisms (SNPs), karyotype, etc., are imported from web services and are available as tracks. In addition, several DAS servers are already included in Genome Maps. As a novelty, this web-based genome browser allows the local upload of huge genomic data files (e.g., VCF or BAM) that can be dynamically visualized in real time at the client side, thus facilitating the management of medical data affected by privacy restrictions. Finally, Genome Maps can easily be integrated into any web application by including only a few lines of code.

Implementation
Genome maps is the official genome viewer of the ICGC and it is implemented in its analysis portal (ICGC Data Portal)
Genome Maps is an open source collaborative initiative available in the GitHub repository.
Genome Maps is available at: http://www.genomemaps.org.
Genome Maps wiki site in GitHub.
Projects using Genome Maps technology out there:
- The Genome Browser of ICGC project (https://dcc.icgc.org/search/g)
- Lens PatSeq Explorer (http://www.lens.org/lens/bio/patseqexplorer)
- Chaetomium thermophilum genome at EMBL (http://ct.bork.embl.de/ctbrowser)
Publication
BiERapp: A web-based interactive framework to assist in the prioritization of disease candidate genes in whole exome sequencing studies

Whole-exome sequencing (WES) has become a fundamental tool for the discovery of disease-related genes of familial diseases and the identification of somatic driver variants in cancer. However, finding the causal mutation among the enormous background of individual variability in a small number of samples is still a big challenge. Here we describe a web-based tool, BiERapp, which efficiently helps in the identification of causative variants in family and sporadic genetic diseases. The program reads lists of predicted variants (nucleotide substitutions and indels) in affected individuals or tumor samples and controls. In family studies, different modes of inheritance can easily be defined to filter out variants that do not segregate with the disease along the family. Moreover, BiERapp integrates additional information such as allelic frequencies in the general population and the most popular damaging scores to further narrow down the number of putative variants in successive filtering steps. BiERapp provides an interactive and user-friendly interface that implements the filtering strategy used in the context of a large-scale genomic project carried out by the Spanish Network for Research, in Rare Diseases (CIBERER) and the Medical Genome Project. in which more than 800 exomes have been analyzed.
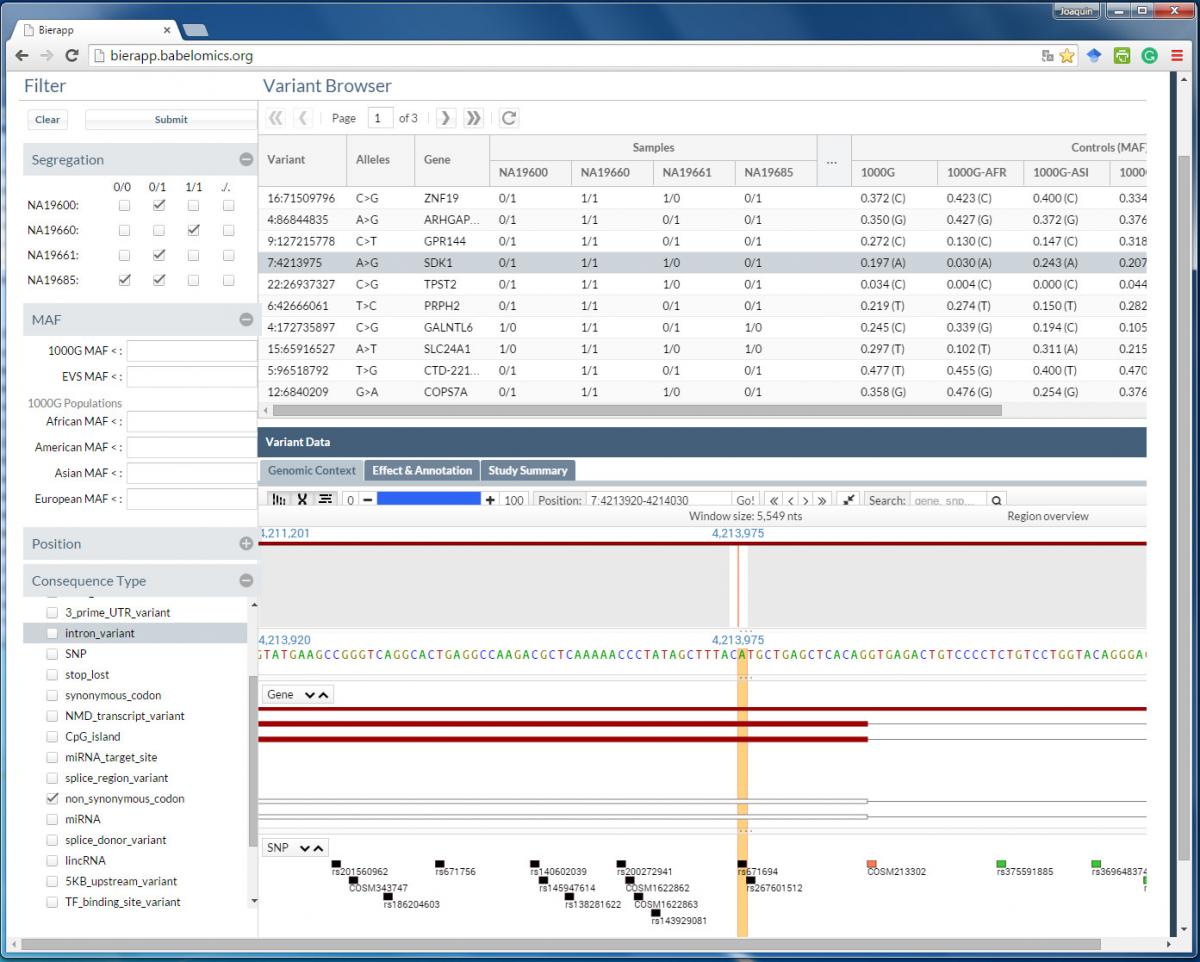
Implementation
BiERapp can be found at: http://bierapp.babelomics.org/
Publication
Alemán A, Garcia-Garcia F, Salavert F, Medina I, Dopazo J. 2014. A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 42:W88-W93
TEAM: A web tool for the design and management of panels of genes for targeted enrichment and massive sequencing for clinical applications
Disease targeted sequencing is gaining importance as a powerful and cost-effective application of high throughput sequencing technologies to the diagnosis. However, the lack of proper tools to process the data hinders its extensive adoption. Here we present TEAM, an intuitive and easy-to-use web tool that fills the gap between the predicted mutations and the final diagnostic in targeted enrichment sequencing analysis. The tool searches for known diagnostic mutations, corresponding to a disease panel, among the predicted patient’s variants. Diagnostic variants for the disease are taken from four databases of disease-related variants (HGMD-public, HUMSAVAR , ClinVar and COSMIC.) If no primary diagnostic variant is found, then a list of secondary findings that can help to establish a diagnostic is produced. TEAM also provides with an interface for the definition of and customization of panels, by means of which, genes and mutations can be added or discarded to adjust panel definitions.
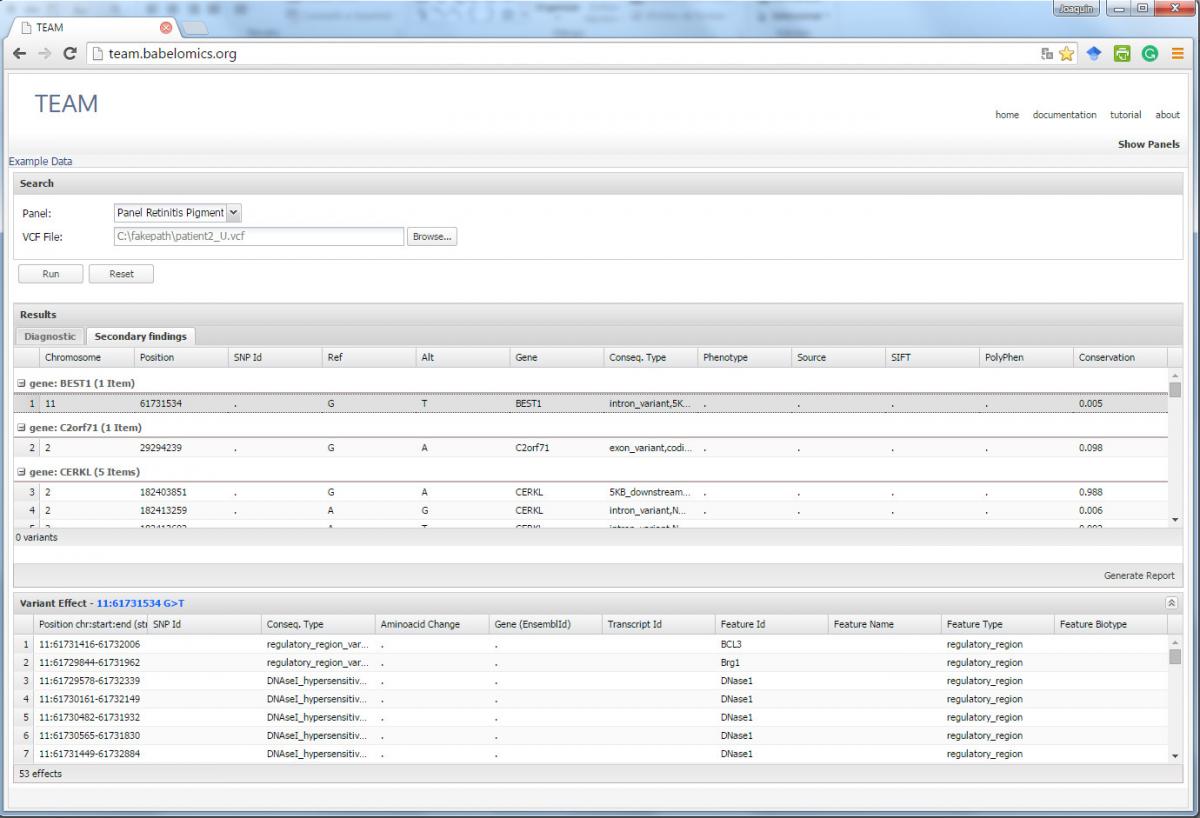
Implementation
TEAM is freely available at: http://team.babelomics.org
Publication
Alemán A, Garcia-Garcia F, Medina I, Dopazo J. 2014. A web tool for the design and management of panels of genes for targeted enrichment and massive sequencing for clinical applications. Nucleic Acids Res. 42:W83-W87
Transcriptional networks

RENATO: Inferring the regulatory network behind a gene expression experiment
Transcription factors (TFs) and microRNAs (miRNAs) are the most important dynamic regulators in the control of gene expression in multicellular organisms. These regulatory elements play crucial roles in development, cell cycling and cell signaling, and they have also been associated with many diseases. The RENATO (REgulatory Network Analysis TOol) web server makes the exploration of regulatory networks easy, enabling a better understanding of functional modularity and network integrity under specific perturbations. RENATO is suitable for the analysis of the result of expression profiling experiments. The program analyzes lists of genes and search for the regulators compatible with its activation or deactivation. Tests of single enrichment or gene set enrichment allow the selection of the subset of TFs or miRNAs significantly involved in the regulation of the query genes. RENATO also offers an interactive advanced graphical interface that allows exploring the regulatory network found.
Implementation
RENATO is available at: http://renato.bioinfo.cipf.es/
Publication
Network Analysis
SNOW: Studying Networks in the Omic World
SNOW stands for "Studying Networks in the Omic World" and complements tools such as FatiGO and Marmite (Babelomics suite) introducing a new dimension in the functional profiling of high-throughput experiments results, this is, protein-protein interaction data. SNOW extracts and evaluates the cooperative behavior of lists of selected proteins/genes using the interactome as scaffold.
SNOW identifies hubs and evaluates the global degree of connections, centrality and neighborhood aggregation of the list by comparing the distributions of nodes connections degree, betweenness centrality and clustering coefficient respectively against the complete distribution of these parameters into the interactome of reference. Besides this, SNOW extracts the minimum network that connects the proteins/genes in the list. A user-fixed number of external proteins to connect nodes in the list is allowed. The topology of this network is evaluated by comparing distributions of graph parameters of this network against pre-calculated distributions of a set (10000) of random lists with same size range. By this, SNOW extracts information about whether the network represented in the list has more hubs, is more connected or has a more regular connections distribution than a random network.SNOW also provides an interactive visualization of the network and a complete description of interatomic and local network parameters of each protein/gene in the list as well as the external nodes introduced by the program. This information together with the functional annotation provided will guide the user to identify the important nodes within, or even outside, the list as well as evaluate the modular functionality of the list as an entity.

More info at: http://snow.bioinfo.cipf.es
Publication
Minguez P, Gotz S, Montaner D, Al-Shahrour F, Dopazo J. 2009. SNOW, a web-based tool for the statistical analysis of protein-protein interaction networks. Nucl. Acids Res.; 37:W109-114
Network miner: Discovering the hidden sub-network component in a ranked list of genes or proteins derived from genomic experiments
Genomic experiments (e.g., differential gene expression, SNP association, etc.) typically produce ranked list of genes. We present a simple but powerful approach which uses protein-protein interaction data to detect sub-networks within such ranked lists of genes or proteins.
We performed an exhaustive study of network parameters that allowed us concluding that the average number of components and the average number of nodes per component are the parameters that best discriminate between real and random networks. A novel aspect that increases the efficiency of this strategy in finding sub-networks is that, in addition to direct connections, also connections mediated by intermediate nodes are considered to build up the sub-networks. The possibility of using of such intermediate nodes makes this approach more robust to noise. It also overcomes some intrinsic limitations intrinsic to experimental designs based in differential expression, in which some nodes are invariant across conditions. The proposed approach can also be used for candidate disease-gene prioritization.
An efficient and easy-to-use web interface (available at http://www.babelomics.org) based on HTML5 technologies is also provided to run the algorithm and represent the network.
Publication
Pathway analysis
PATHiWAYS: Inferring the functional effect of gene expression changes in signaling pathways
Signaling pathways constitute a valuable source of information that allows interpreting the way in which alterations in gene activities affect to particular cell functionalities. There are web tools available that allow viewing and editing pathways as well as representing experimental data on them. However, very few methods aimed to identify the signaling circuits, within a pathway, associated to the biological problem studied exist and none of them provide a convenient graphical web interface. We present PATHiWAYS, a web-based signaling pathway visualization system that infer changes in signaling that affect cell functionality from the measurements of gene expression values in typical expression microarray case-control experiments. A simple probabilistic model of the pathway is used to estimate the probabilities for signal transmission from any receptor to any final effector molecule (taking into account the pathway topology) using for this the individual probabilities of gene product presence/absence inferred from gene expression values. Significant changes in these probabilities allow linking different cell functionalities triggered by the pathway to the biological problem studied.
PATHiWAYS is available at: http://pathiways.babelomics.org/
Publication
Sebastián-León P, Carbonell J, Salavert F, Sanchez R, Medina I, Dopazo J. 2013. Inferring the functional effect of gene expression changes in signaling pathways. Nucleic Acids Res. 41:W213-W217
Sebastian-Leon P, Vidal E, Minguez P, et al. 2014. Understanding disease mechanisms with models of signaling pathway activities. BMC Syst Biol. 8(1):121
GenomeWeb quoted PATHiWAYS.
PATHiVar: Assessing the impact of mutations found in next generation sequencing data over human signaling pathways
Modern sequencing technologies produce increasingly detailed data on genomic variation. However, conventional methods for relating either individual variants or mutated genes to phenotypes present known limitations given the complex, multigenic nature of many diseases or traits. Here we present PATHiVar, a web-based tool that integrates genomic variation data with gene expression tissue information. PATHiVar constitutes a new generation of genomic data analysis methods that allow studying variants found in next-generation sequencing experiment in the context of signaling pathways. Simple Boolean models of pathways provide detailed descriptions of the impact of mutations in cell functionality so as, recurrences in functionality failures can easily be related to diseases, even if they are produced by mutations in different genes. Patterns of changes in signal transmission circuits, often unpredictable from individual genes mutated, correspond to patterns of affected functionalities that can be related to complex traits such as disease progression, drug response, etc.

PATHiVar is available at: http://pathivar.babelomics.org/
Publication
Hernansaiz-Ballesteros RD, Salavert F, Sebastián-León P, Alemán A, Medina I, Dopazo J. 2015. Assessing the impact of mutations found in next generation sequencing data over human signaling pathways. Nucleic Acids Res. 43 (W1): W270-W275.
High performance genomics
HPG Aligner
HPG Aligner applies suffix arrays and/or Burrows-Wheeler transform for NGS read mapping (DNA or RNA). This implementation produces a highly sensitive and extremely fast mapping of reads that scales up almost linearly with read length. The approach presented here is faster (over 20× for long reads) and more sensitive (over 98% in a wide range of read lengths) than the current state-of-the-art mappers. HPG Aligner is not only an optimal alternative for current sequencers but also the only solution available to cope with longer reads and growing throughputs produced by forthcoming sequencing technologies.
HPG Aligner is available in GitHub at http://github.com/opencb/hpg-aligner
HPG Pore
HPG Pore is a toolkit for exploring and analysing nanopore sequencing data. HPG Pore can run on both individual computers and in the Hadoop distributed computing framework, which allows easy scale-up to manage the large amounts of data expected to result from extensive use of nanopore technologies in the future.
The use of nanopore technologies is expected to spread in the future because they are portable and can sequence long fragments of DNA molecules without prior amplification. The first nanopore sequencer available, the MinION™ from Oxford Nanopore Technologies, is a USB-connected, portable device that allows real-time DNA analysis. In addition, other new instruments are expected to be released soon, which promise to outperform the current short-read technologies in terms of throughput. Despite the flood of data expected from this technology, the data analysis solutions currently available are only designed to manage small projects and are not scalable.
Implementation
HPG Pore is available in GitHub at http://github.com/opencb/hpg-pore
ngsCAT
ngsCAT (Next Generation Sequencing data Capture Assessment Tool) is a command-line application written in Python which facilitates a comprehensive evaluation of the performance of the capture step in targeted high-throughput sequencing experiments in terms of:
-
Sensitivity, which assesses the quality of the coverage on target regions. It is also important to provide a means of estimating how this coverage would improve by increasing sequencing depth.
- Specificity, which measures how much of the sequencing effort is wasted on sequencing off-target bases.
-
Uniformity, which assesses sequencing biases due to specific genomic locations or nucleotide composition.
ngsCAT is an easy-to-use tool that can be run with just one command line in a standar computer, generating a detailed HTML report with metrics, summary tables, figures and plots that evaluate the efficiency of targeted enrichment sequencing.
Implementation
ngsCAT is availabl at ngsCAT.
Packages
Babelomics: advanced functional profiling of transcriptomics, proteomics and genomics experiments
Babelomics has been running for more than one decade offering a user-friendly interface for the functional analysis of gene expression and genomic data. Here we present its fifth release, which includes support for Next Generation Sequencing data including gene expression (RNA-seq), exome or genome resequencing. Babelomics has simplified its interface, being now more intuitive. Improved visualization options, such as a genome viewer as well as an interactive network viewer, have been implemented. New technical enhancements at both, client and server sides, makes the user experience faster and more dynamic. Babelomics offers user-friendly access to a full range of methods that cover: (i) primary data analysis, (ii) a variety of tests for different experimental designs and (iii) different enrichment and network analysis algorithms for the interpretation of the results of such tests in the proper functional context.
Babelomics contains one of the most cited tools in the field of functional enrichment analysis, the FatiGO. See quotation in ScienceWatch.

Implementation
Babelomics is available at http://www.babelomics.org
Babelomics Tutorial
Publication
A full chapter is dedicated to Babelomics in the popular Molecular Cloning textbook as a protocol for gene expression data analysis
Al-Shahrour F., Minguez P., Vaquerizas J.M., Conde L., Dopazo J. 2005. BABELOMICS: a suite of web tools for functional annotation and analysis of groups of genes in high-throughput experiments. Nucleic Acids Res. 33:W460-W464
Al-Shahrour F., Minguez P., Tarraga J., Montaner D., Alloza E., Vaquerizas J.M., Conde L., Blaschke C., Vera J., Dopazo J. 2006. BABELOMICS: a systems biology perspective in the functional annotation of genome-scale experiments. Nucleic Acids Res. 34:W472-W476
Al-Shahrour F, Carbonell J, Minguez P, Goetz S, Conesa A, Tarraga J, Medina I, Alloza E, Montaner D, Dopazo J. 2008. Babelomics: advanced functional profiling of transcriptomics, proteomics and genomics experiments. Nucleic Acids Res. 36:W341-346
Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tárraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, García F, Marbà M, Montaner D, Dopazo J. 2010 Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Research. 38:W210-W213.
Alonso R, Salavert F, Garcia-Garcia F, Carbonell-Caballero J, Bleda M, Garcia-Alonso L, Sanchis-Juan A, Perez-Gil D, Marin-Garcia P, Sanchez R, Cubuk C, Hidalgo MR, Amadoz A, Hernansaiz-Ballesteros RD, Alemán A, Tarraga J, Montaner D, Medina I, Dopazo J. 2015. Babelomics 5.0: functional interpretation for new generations of genomic data. Nucleic Acids Res. 43:W117-W121
Related papers:
Al-Shahrour F, Díaz-Uriarte R, and Dopazo J, 2004, FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 20(4):578-580.
Medina I, Montaner D, Tarraga J and Dopazo J, 2007, Prophet, a web-based tool for class prediction using microarray data. Bioinformatics 23 (3): 390-391.
Al-Shahrour F, Minguez P, Tarraga J, Medina I, Alloza E, Montaner D and Dopazo J. 2007.FatiGO +: a functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucl.Acids Res. 35: W91-W96.
GEPAS: A web-based tool for microarray data analysis and interpretation
GEPAS was one of the most complete and extensively used web-based packages for microarray data analysis. During its more than 10 years of activity it has continuously been updated to keep pace with the state-of-the-art in the changing microarray data analysis arena. In 2013 GEPAS was discontinued and all its functionality was implemented in Babelomics.
GEPAS offered diverse analysis options that included well established as well as novel algorithms for normalization, gene selection, class prediction, clustering and functional profiling of the experiment. New options for time-course (or dose-response) experiments, microarray-based class prediction, new clustering methods and new tests for differential expression wer included in subsequent versions.
An extensive re-engineering of GEPAS was carried out which included the use of web services and Web 2.0 technology features, a new user interface with persistent sessions and a new extended database of gene identifiers. GEPAS has been the most quoted web tool in its field and it has been extensively used by researchers of many countries. GEPAS statistics indicate an average usage rate of 500 experiments per day.
All the functionality of GEPAS, is now available within Babelomics http://www.babelomics.org.
Tools no longer supported
Pupasuite: SNP prioritization for large scale genotyping and functional characterization of SNPs in next generation sequencing experiments
PupaSuite is a web tool for the selection of SNPs with potential phenotypic effect, oriented to help in the design of large-scale genotyping projects and to the characterization of new SNPs from next generation technologies.
PupaSuite uses a collection of data on SNPs from heterogeneous sources and a large number of pre-calculated predictions to offer a flexible and intuitive interface for selecting an optimal set of SNPs. It implements new facilities such as the analysis of user's data to derive haplotypes with functional information. A new estimator of putative effect of polymorphisms has been included that uses evolutionary information. Also SNPeffect database predictions have been included.
PupaSuite 2 can input lists of SNPs, genes, or chromosomal regions and produce lists of SNPs with their possible phenotypic effect. Moreover, it can also calculate haplotypes and estimate the possible functionality of the corresponding combinations of SNPs.
PupaSuite can also produce graphic interactive analyses of single genes. There, a filter allows displaying SNPs potentially causing different types (structural, regulatory, splicing, etc.) and degrees of alterations.

Phylemon: a suite of web-tools for molecular evolution, phylogenetics, phylogenomics and hypothesis testing
Phylemon 2.0 is the second release of the suite of web-tools for molecular evolution, phylogenetics and phylogenomics. It is conceived as a natural response to the increasing demand of data analysis of experimental scientists seeking to add molecular evolution and phylogenetic insight into their research.
Phylemon 2.0 has several features that differentiate it from other web resources: (i) It offers an integrated environment that enables the direct concatenation of evolutionary analyses, format conversions, the storage and edition of projects and results,(ii) Phylemon suggests the next possible analyses, thus guiding the user and facilitating the integration of multi-step analyses, and (iii) users can define and save pipelines for specific phylogenetic analysis to be used on many genes in subsequent sessions or multiple genes in a single session (phylogenomics).
The new web server integrates a suite of more than 30 different tools such as sequence alignment of long genomic regions, alignment refinement, tree reconstruction by distances using the best DNA model explaining the data, DNA model selection and averaging using the jModelTest and HyPhy tools to test for molecular adaptation considering synonymous rate variation across sites. Phylemon 2.0 has been completely re-engineered improving job submission, project, file, and task management.

more info at: http://phylemon.bioinfo.cipf.es